生物等效性试验中受试者例数的确定(全)
发稿时间:2017-01-25 来源: 培优创新论坛
问:在仿制药一致性评价生物等效性试验中,如何计算样本量或受试者的例数?
答:对于这个问题,首先要分清楚仿制药本身是普通口服固体药物、高变异度的药物还是窄治疗指数的药物。不同类别的药物计算方法略有不同。
普通口服固体药物
1.例数要满足统计学要求,其次要符合法规要求。
2016年国家食品药品监管总局(CFDA)《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》要求:入选受试者的例数应使生物等效性评价具有足够的统计学效力。2015年《药物制剂人体生物利用度和生物等效性试验指导原则》中指出,应该根据适当的样本量计算法,确定包括在试验中的受试者数目。在一项生物等效性试验中,可评价的受试者数目不应少于18 名。
2.计算方法有查表法、公式法和软件法,所得结果基本一致,但需要注明计算公式、表格出处或用的是哪种软件。
公式法:在显著性水平α达到(1-β)把握度所需的受试者例数计算公式如图1,n为每个序列受试者的例数;两周期交叉试验所需总受试者例数N=2n;CV为个体内变异系数。
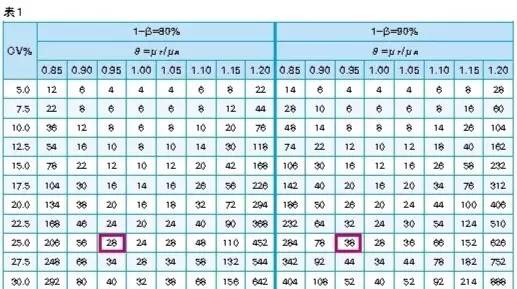
查表法:在α=0.05,相同的CV时,要达到较高的把握度,所需要的受试者的例数越多,因此要达到80%和90%的把握度,所需受试者例数N也有区别(见表1):
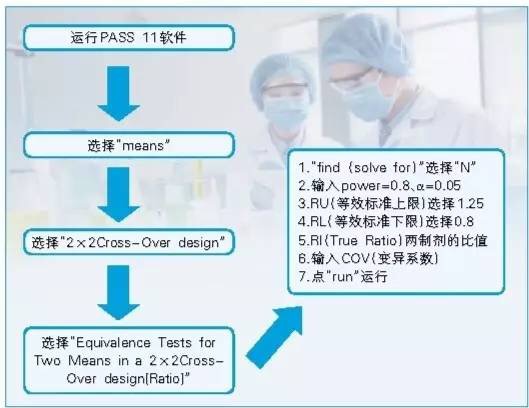
软件法:PASS、nQuery、S+和SAS等软件都可以计算生物等效性试验的样本量。
样本含量估计以药代动力学参数的变异系数为计算指标,α=0.05,1-β=0.8,等效标准为80.00%~125.00% ,双交叉设计试验的计算过程如图2。
软件法和查表法的比较:从表2中可以看出,当变异系数相等,α=0.05,把握度为80%时,软件法和查表法计算结果基本一致。
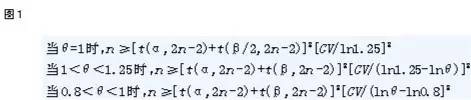
高变异性药物
高变异性药物是指药动学参数个体内变异大于等于30%的药品。
对于高变异药物生物等效性试验,可采用参比制剂校正的平均生物等效性(RSABE)方法,该方法可根据参比制剂的个体内变异性对等效限的判定范围进行比例化的调整。
不同的国家法规在药代动力学参数(Cmax,AUC) 90%CI的等效性判定标准方面略有差异。CFDA的指导原则明确指出:对于高变异药物,可根据参比制剂的个体内变异,将等效性评价标准作适当比例的调整,但调整应有充分的依据。如果认为Cmax差异较大,对于临床的影响不大,基于临床的充分理由,则可以放宽接受范围。在这种情况下,Cmax的接受范围可以最宽为69.84%~143.19%, AUC的接受限保持在80.00%~125.00%。
若采用RSABE方法对等效限范围进行放宽,要求对参比制剂重复设计,包括部分重复和完全重复设计,或可采用不放宽等效限范围、仅增加受试者例数的方法。
在计算样本量前,需要获得个体内变异。参比制剂的个体内变异系数可通过以下几种方法获得:查阅公开数据报道;进行一个小规模预试验,获得该药物准确可靠的个体内变异,然后根据该数据进行受试者例数的估算。
窄治疗指数药物
窄治疗指数药物是指剂量或血药浓度的微小变化可能导致严重的治疗失败和/或严重的不良反应,危及生命或引起持续或明显的残疾或功能不全的药物。2015年《药物制剂人体生物利用度和生物等效性试验指导原则》中指出:对于治疗指数窄的药品的特殊情况,AUC 的可接受区间应该被缩窄为90.00%~111.11%。在Cmax对安全性、药效或药物浓度监测特别重要的情况,该参数也应适用90.00%~111.11%的接受限。因此在计算该类药物的受试者例数时,要注意等效范围的改变。
在计算生物等效性受试者的例数时,还需注意以下几点:计算受试者例数采用的CV是参比制剂的个体内变异CV;对于高变异药物、窄治疗指数药物进行生物等效性研究时,如果采用RSABE方法,对等效限进行放宽的,建议采用EMA的标准设计方案;如果采用RSABE方法,研究设计必须重复交叉设计试验,FDA推荐RSABE研究不少于24例。
(本文转载自培优创新论坛)