发稿时间:2019-02-16 来源:CDE
摘要:抗肿瘤药物的Ⅱ期临床试验主要目的是在预先确定的某个患者群体中探索药物的临床活性或生物学活性,提供令人信服的证据,以吸引来自各方面的对III期试验的投入。分子靶向药物的开发对传统的抗肿瘤药物早期临床研究设计提出了挑战,许多新的研究策略有助于及早筛选出有效药物进入大样本的III期临床试验。本文概括介绍了Ⅱ期临床试验设计中的基本要点包括药物的选择、剂量的选择、研究人群的选择、评价终点的选择等,以及对于分子靶向药物与这些要点有关的特殊考虑。同时也介绍正在探索中的一些新的Ⅱ期临床试验设计方法。内容摘自KeithT. Flaherty, MD和Peter J. O’Dwyer,MD发表并收录在《Cancer Drug Discovery andDevelopment》一书中的文章,期望能对正在从事抗肿瘤药物临床研究工作的相关人员提供参考。
1. 前言
肿瘤药物的早期临床研究不同于其它临床学科的治疗评价。首先,用于治疗肿瘤性疾病的药物的相关危险性决定了这类药物只能在肿瘤患者中进行评价,而不能在健康志愿者中进行评价(目前学术界对于毒性低的靶向治疗药是否可用健康志愿者进行I期临床有不同观点和做法-译者注)。第二,量效关系的评价对于了解药物的潜在治疗受益和毒性作用是必不可少的。第三,要研发出有效的治疗方案,必须明确该方案对某些亚群患者群体有独特的治疗价值。基于上述原因,目前已建立了一套合理的评价体系,对可耐受并且有效的治疗方案进行优化选择,以便在随后的临床试验中和标准治疗方案进行比较。
进行I期临床试验的目的是为II期研究确定推荐剂量和给药方案。I期临床试验最主要的目标是确定不良反应的发生率、严重程度和可逆性。这些结果主要取决于试验药物在不完全相同患者人群中的体内分布、代谢和消除情况。药代动力学研究可以对具有遗传多样性的人群中药物暴露的变异情况进行量化分析。而变异的程度是该药物是否适合进行进一步评价的决定因素。将药代动力学与毒理学研究相结合,可以对给药方案做出选择。这些研究一般是在具有不同治疗背景的少量患者中进行。为了方便起见,各种各样的癌症患者都要入选。即使在有着相同的既往治疗史和基线器官功能的患者中,药物代谢和消除也存在个体差异。这些个体差异造成的风险是找不到恰当的II期给药剂量。这种情况下,进行II期试验可进一步调整后续试验所需的剂量。
药物和宿主之间的相互作用决定了药物的药代动力学。这一点必须与药物和它的分子靶位之间的相互作用以及其后续效应区别开来,这种后续效应被称为药效学作用。I期试验的重点应侧重在药代动力学上,而II期试验,重点则转移到药效学上。也就是说,药物开发中的首要目标是要在某个患者人群中建立体内过程相对比较一致且药物的毒性可以接受的一种试验疗法。但是,I期试验可以对药效检测方法的可行性和再现性进行探索。
II期临床试验的主要观察指标是在预先确定的某个患者群体中探索药物的临床活性或生物学活性。临床活性的定义和用于评价这种活性的方法近年来一直在进行重大修改。在分子靶向治疗的时代,生物学活性必然要与临床受益结合起来。如果I期临床试验设定了药效学终点的观察,那么就可以确定生物学活性的阈剂量。阈剂量可以大大低于最大耐受剂量,而最大耐受剂量一直是传统I期试验的终止剂量。II期临床试验可以设计成同时评价一个相对临床疗效较低的生物学有效剂量以及最大耐受剂量的研究。最终,II期研究必须提供令人信服的证据,以吸引来自各方面的对III期试验的投入。对于一个有独特活性的药物,如果可以明确其疗效优于标准治疗方案,则可以得到FDA的批准。最近被批准用于慢性髓性白血病和胃肠道间质瘤的药物STI571(Sunitinib,Pfizer)就是一个实例。
2. II期临床试验的基本要点
2.1. 药物选择
进行I期、II期和III期临床试验的费用一般分别为20万美元、100万美元和1000万美元。通常情况下,一种新的抗肿瘤药物要在1-4个I期试验中对不同的方案进行评价。II期临床试验将在5-10个肿瘤类型中进行。从I期到II期临床试验的投资需要增加10倍。由于对临床研究的公共资助和私人资助是有限的,所以,这就要求用于II期临床开发的药物必须经过严格的选择。
对开展II期临床试验的药物进行筛选的标准还在不断地完善。传统的细胞毒类药物有非常强的剂量依赖性治疗作用和剂量依赖性毒性。治疗指数窄是I期试验后放弃一种药物的原因之一。并且,毒性必须是可以治疗和具有可逆性,这样才可以用于姑息治疗。除此之外,还要求药物清除的个体间变异应当非常小,一般应低于3倍。最后,毒性应直接与药物的暴露量成正比,并可以预测。在动物模型中分子靶向治疗的治疗指数似乎比较高,且不受剂量限制性毒性发生率的限制。在人体中诱导出预期的分子效应是筛选进行II期开发的分子靶向治疗方案的新标准。

2.2. 剂量调整
虽然I期试验的主要作用是明确安全剂量和给药方案,但是,由于既往治疗史不同、样本量的限制以及不能检测出累积毒性等,推荐的II期用药剂量可能不能推广到比较大规模的人群。如果入选I期临床研究的大多数患者已经接受过多种骨髓抑制的化疗方案,那么他们与那些在II期研究中用试验药物作为一线治疗的患者相比,对试验药物的骨髓抑制作用更加敏感。这类错误可能导致试验药物的用药剂量不足。I期研究中治疗的患者例数相对比较少,因此没有足够的把握能检出不常见的但为重度的毒性。这种情况下,推荐的II期用药剂量可能对耐受剂量估计过高。最后,被选入I期研究的患者一般都是标准治疗无效的癌症患者,由于病情进展,他们不可能在I期试验中接受长期治疗。如果多数患者在前几周内停止治疗,由于样本量小,所以有关累积毒性的数据就不可靠。
II期试验可以在研究过程中调整试验药物的剂量从而克服这些错误。可以预先确定规则,在重度毒性的发生率过高,不能耐受的情况下,可以降低药物的起始剂量。例如,4级骨髓抑制的发生率如果为33%,一般认为是不能接受的。可以制定停药的规则,即,如果在最初的患者队列中,有一定数量的患者发生4级骨髓抑制,就可以根据规则,在以后的患者中降低药物的起始剂量。这种统计学处理方式一般用于疗效,但同样也可以用于毒性(表1)。
I期试验中得到的药代动力学数据也可用于根据药物清除情况的差异进行剂量调整。在卡铂的开发中就采用了这种方法。卡铂的清除率与肌酐清除率之间有非常好的相关性(1),因此建立了一个模型,即根据目标的药时曲线下面积、体表面积和肌酐清除率来确定合适的剂量。与单纯根据体表面积决定给药剂量的方法相比,用这种方法,可显著降低肾衰和重度骨髓抑制的危险性。
2.3.治疗组的选择
2.3.1. 患者的特定因素
已经证明了,几个与患者相关的因素对于II期试验的毒性和临床反应的准确评价有重要作用。对化疗药物的前期暴露情况是后续治疗中某些副作用的重要预测因素(2)。由于(在)既往治疗中的药物暴露所引发的耐药机制可能造成对试验药物的重叠耐药。所以,与那些既往用药不多的患者相比,就可能导致所预期的药物疗效明显降低。在以前使用过多柔比星治疗的乳腺癌妇女中进行米托蒽醌的研究时,其评价就遇到了这种情况(3,4)。药物的早期研究中需要只纳入器官功能正常的患者,因为器官功能不正常时清除率就可能受到影响,从而导致药物的暴露量增加,药物毒性过高等情况。
如果选择既往治疗有限的患者参加试验,可能有重要的伦理学问题,也就是试验方案在癌症患者治疗规则中的定位问题。普遍认为如果有治愈性治疗方案存在的情况下,患者应当采用标准治疗方案作为一线治疗。复发的时候,患者才能参加试验药物的II期试验。对于将化疗仅仅作为姑息治疗的疾病,什么样的治疗先后次序比较合理,目前还有争论。就转移性乳腺癌来说,不治疗的情况下,从诊断出转移癌的时间算起,患者的中位生存期大于2-1/2年。序贯治疗可以将中位生存期延长到4年以上。虽然存在几种治疗乳腺癌的有效药物,但还没有证据显示在整个治疗过程早期应用一种试验药物对总的生存期有何负面影响。并且,为了评价试验药物与当前一线治疗相比的潜在优效性,有必要在II期试验中估计一线治疗时试验药物的活性。目前还不太清楚在疾病本身不需要多种序贯治疗的患者中,如何安排标准治疗和试验治疗的先后次序。
体力状态是细胞毒类药物毒性增加和临床受益降低的独立危险因素。两个经过验证的量表已明确了禁止化疗的体力状态水平,这两个量表是东部合作肿瘤组(ECOG)量表(5)和Karnofsky量表(6)。这些量表很容易应用于一般的医疗实践中,具有高度的可重复性。这些量表是疾病进展及其生理学影响的客观测量指标。一般情况下,ECOG体力状态为0或1的患者适合入选II期试验。对于一些新药来说,其副作用可足以被耐受,体力状态为2的患者也可以入选。
高龄不是入选II期试验的强制性限制条件,细胞毒类药物的许多副作用未观察到年龄依赖性。对于和年龄相关的副作用,通过对器官功能的仔细评估,常常可以发现患者的药物清除率的降低。也有一些药物,其严重的副作用在较年轻的患者中更常见(7),而另外一些药物则相反(8,9)。目前已知有一些药物在年龄非常小的儿童中,其药代动力学特性与各个年龄段的成人有明显不同(10,11)。因此,往往要在儿童中进行单独的剂量探索和I期试验。一般认为,非激素治疗不存在性别差异。但是,由于激素对细胞色素P450氧化还原酶系统的影响,有些药物的代谢可能存在不同(12)。同样,虽然在入选条件上不会对种族加以限制,但种族差异也与P450酶的基因型和表型差异有关(13,14)。
II期临床试验中的单药疗效可以与在类似患者中的标准治疗的疗效进行比较,但可能会受到研究人群中存在的混杂因素的影响,这些混杂因素可能导致过低或过高估计临床受益情况。尽管如此,II期试验中临床疗效的评估结果仍然是进行随机化III期试验的基础。
2.3.2. 肿瘤的特定因素
一般选择II期试验目标人群的策略是选择肿瘤病变具有共同组织学亚型和共同起源部位的患者。这种入选方法具有两个优点:一是结果可以归纳总结,二是发现预期之外的作用机制。
对II期试验中的目标疾病的合理选择,常因体外系统和动物模型的预测性不好而受到限制。一般对试验药物的疗效初筛,通常选择标准的细胞毒化疗方法治疗有效的恶性肿瘤或病变(15)。但在传统的细胞毒类药物或分子靶向治疗应用中,这种方法不能发现药物对非代表性肿瘤的疗效,并且根据临床前的结果,导致在II期试验中投入过多的资源。这种情况已被非经验性的方法所取代,即将人的肿瘤细胞移植到小鼠来选择肿瘤类型。但是,这种方法也会出现不好的阳性和阴性预测值(16)。对广谱的人实体瘤细胞株的体外活性已成为选择目标疾病的更特异的方法,但单独使用也不能准确预测药物在II期试验中的疗效(17,18,19)。
回顾过去的研究,进一步的工作已经阐明了这些模型不能代表人类肿瘤的某些原因。小鼠的人肿瘤移植模型只能在免疫受损的宿主中取得成功,因此,这个系统不能诱导继发的免疫反应,而后者是既往被忽略的许多有效的细胞毒类药物的作用机制。并且,由于移植部位的生理条件不同,移植瘤的生长会导致与人体自身肿瘤所不同的肿瘤基质和微血管系统的出现。这对于血管生成抑制剂的疗效特别有意义。据报道许多细胞毒类药物也有抗血管生成的活性,因此也可以解释临床前活性和临床疗效之间的差别。根据早期对针对发病机制中重要靶点的特异性药物的研发经验表明,联合应用靶点依赖的细胞株和相应人肿瘤细胞株的小鼠移植瘤,可更好地发现有效的药物。
单纯根据病理学表型入选患者还可以对药物的作用机制进行假设检验。对于分子靶点已经明确的药物,常常假设其临床疗效局限于肿瘤过度表达或过度利用这种分子靶点的患者。这一假说的根据是药物对某个功能已经得到充分认识的靶点具有选择性。有关表皮生长因子(EGF)受体(erb-b1)小分子抑制剂的临床前研究提示,其抗增生活性仅限于该受体过度表达的肿瘤类型。但是,数个动物模型研究发现,该受体没有过度表达的肿瘤类型中,也可以诱导出肿瘤缓解。这种疗效与药物对血管生成的抑制作用有关。
根据共同的分子表型入选患者是II期试验的另一策略。目前已有数个例子成功确立了预测靶向治疗疗效的标记物。c-erb-2单克隆抗体曲妥珠单抗(TRASTUZUMAB,Roche)对转移性乳腺癌有显著疗效,基因扩增方法显示30%的乳腺癌患者有c-erb-2受体的过表达,是否存在c-erb-2基因扩增可以准确预测出能从该项治疗中获得临床受益的患者。这充分说明了确立分子表型检测方法的重要性。应用免疫组化方法可以对c-erb-2蛋白的过表达进行半定量检测,但与基因扩增相比,前者显然不够精确,会错误判定病人是否存在c-erb-2过表达。在进行个体化靶向治疗中,这种错误会导致选择治疗方案中的关键性失误。
DNA突变状态和基因表达谱为II期试验提供了另外一个分子水平的筛选方法。研究发现2/3有转移性黑色素瘤的患者存在BRAF激酶域体细胞突变。这些突变基因的表达导致b-raf激酶持续活化,通过促分裂原活化蛋白(MAp)激酶通路的信号传导促进细胞存活。Raf激酶的小分子抑制剂BAY43-9006治疗转移性黑色素瘤的II期试验正在进行中。可以根据BRAF突变来筛选患者,但是,对突变的错误分类和存在其他作用机制可能会排除部分有疗效的患者。事实上,抗血管生成似乎是raf信号传导抑制剂的一个重要作用机制。因此,恰当的筛选策略应当是入选所有转移性黑色素瘤的患者,收集所有患者BRAF突变的信息,以便明确突变与临床受益的相关性。
虽然基因表达谱不能鉴别是否存在一个特异性的分子靶点,但它可以从分子水平上根据肿瘤的生物学特性对病人进行分类,从而将表型与疗效联系起来,以便在随后的III期试验中根据上述预测方法对患者进行分层。在套细胞淋巴瘤(mantle cell lymphoma)患者中,两个不同的表达谱型可以区分生发中心型和滤泡型肿瘤。这一分类可提供重要的预后信息,帮助识别标准治疗无效的患者,从而使这一组患者可以免受无效治疗的毒性,直接进入新治疗方案的临床试验。
2.4. 确定终点
2.4.1. 临床终点
延长生存期是抗肿瘤治疗的根本。但是,由于存在混杂因素,在小规模的单臂临床试验中很难准确评估生存受益情况。选择偏倚可以导致无意识地入选一些由于自身因素或肿瘤特征造成疾病生长缓慢或进展迅速的患者。这些因素可能超出治疗作用,导致对疗效的错误估计。如果以总生存期作为治疗受益的唯一指标,由于受到后续治疗对疾病进展的影响,情况会变得更加复杂。这对于生长相对缓慢的肿瘤来说尤其重要,因为这类肿瘤在患者死亡之前可能用过多个疗程的治疗。已经提出将疾病无进展的时间作为一个比较好的临床受益评价指标,因为这个指标不受后续治疗的干扰,特别是对具有抑制细胞生长或抗血管生成作用的药物。在单臂临床试验中,应用这一指标同样存在选择偏倚。
客观缓解率一直是试验药物临床疗效的标准替代指标。因为肿瘤很少出现自发缓解,所以肿瘤缩小被认为是治疗特有的反应。有关治疗后肿瘤大小的评价标准已经确立,但这种评价临床受益的体系也有局限性,就是要求患者存在可以准确测量的内脏肿块。对于骨骼、淋巴结和皮下转移瘤,这种评价方法的作用有限,因此对于前列腺癌等临床表现主要集中在上述组织的疾病,引入了骨痛缓解和血清肿瘤标志物水平降低作为疗效的替代指标。虽然技术上可行,但这些测量指标与肿瘤负菏或随后的总生存期的相关性并不好。并且,对部分肿瘤类型,特别是小细胞肺癌和结直肠癌,部分缓解在预测肿瘤进展延迟或生存期延长中的意义尚不明确。由于上述原因,有人提出了其他的替代终点。
生活质量已经成为II期试验中确定的组成部分,因为这样可较早的对明确的副作用的影响进行量化。由于患者入选时存在选择偏倚,单组II期研究中评价生活质量的意义有限,但在随机化II期研究中评价生活质量则非常有意义。确立标准化的、与操作者无关的、简单易用的评价方法将强化这一终点在试验药物研发中的意义。虽然FDA不会仅仅根据与标准治疗相比生活质量有所改善就批准一种药物,但II期试验中在这一终点上明显处于劣势的药物进行进一步研究的可行性会受到影响。
2.4.2. 药代动力学终点
药代动力学虽然在常规上属于I期临床试验和临床前研究的范畴,但现在已成为药物研发中各阶段的组成部分(20)。由于入选患者的个体差异可以对临床疗效和肿瘤药物浓度产生深刻影响,所以在II期临床试验中继续考察药代动力学和药效学是有益处的。其目的不仅仅是扩大药代动力学-药效学数据以支持所提出的模型,而且也是要明确哪些患者因素或协变量影响了药物的分布和疗效。此类模型的应用不仅可以通过维持目标血药浓度来改善患者的疗效或避免毒性,而且可以增进对药物作用机制的认识,还可以利用II期试验的数据对I期试验所提出的模型进行充分的细化。
把重点放到药代动力学上可以通过应用药代动力学和药效学模型对患者间存在的差异进行量化,并且认识到AUC和稳态浓度等药代动力学参数与治疗疗效和骨髓抑制的相关性优于剂量与这些指标的相关性(21)。有限采样模式(limitedsampleing model)可以通过采集比较少的标本来确定特定患者的药代动力学,这种模式为II期试验数据比较少的情况下,根据药代动力学确定给药方案提供了可行的方法。
虽然计算的工作量比较大,但群体药代动力学分析可以把II期临床试验收集的观察性数据综合起来。这些方法对于区分个体之间、各次试验之间和个体本身来源的药代动力学和药效学变异方面的作用尤为突出,同时还可以解决不平衡和混杂因素等研究设计问题(22,23)。性别、年龄和血清肌酐水平等患者方面的因素对于药物发挥作用也很重要,将这些因素与药代动力学和患者对药物的反应一起进行分析,通过贝叶斯个体化分析与群体分析结果相结合的统计方法,对于特定患者亚群的给药方案做出合理的调整(24)。
研发耐受性更好的靶向治疗方案可以实现传统细胞毒类药物无法实现的给药策略。许多研发中的药物可以长期使用而不出现药物蓄积引发的毒性。虽然对这些药物也必须确定同样的药代动力学参数,但给药方案更多地根据血清半衰期,而不是依据剂量依赖性副作用恢复所需要的时间。选择这些药物用药方案的目标是某个阈值以上的持续暴露量,或者是对分子靶点的持续抑制作用。某些情况下,II期研究的给药方案是依据能够实现动物模型中与药效学作用相关的稳态浓度的I期给药方案制定的。其他情况下,药物浓度可能检测不到,但分子靶点仍然能被抑制较长的时间。无论是哪种情况,II期试验中药代动力学分析都可以进一步探讨药物暴露量和临床疗效的关系。在涉及到药效学终点时,药代动力学模型支持用稳态浓度作为药效反应的替代指标。
2.4.3. 分子终点
分子靶向治疗以及可以对分子靶点的活性进行量化的实验室检测方法的出现,为在体内对试验方案的作用机制进行验证提供了基础(图1)。此类药效学研究必须满足多个技术指标,才能提供有意义的预测信息。首先必须能够对分子靶点进行可靠的检测。对于导致靶点分子表达下调的药物,单纯测量靶点的表达就可以作为药物疗效的预测指标。但是,对于抑制或激活分子靶点的药物,则必须对其活性进行精确的检测。治疗前和治疗中对同一肿瘤组织进行活检可以得到配对的组织标本,从而了解靶点的表达情况或活性。另外,也可以用较易获得的治疗前和治疗中的正常组织代替肿瘤组织。理想情况下,应当在肿瘤组织和替代组织中同时进行药效学研究,这样就可以分析药物对分子活性的作用,并验证替代组织的有效性。如果早期确立了适宜的替代组织,就可以在随后的III期试验中对入选的患者进行前瞻性的药效作用评价。

图1. 靶向治疗的评价模式
目前正在研发中的几个治疗药物都缺乏对靶点抑制作用的验证。ZD1839(Gefitinib,Astrazeneca)是表皮生长因子受体(EGFR)酪氨酸激酶的一种小分子抑制剂,在其I期临床试验中,对部分患者先后多次进行了皮肤活检,以获得替代组织。EGFR酪氨酸激酶的激活导致受体的自身磷酸化,目前已研发出了可以识别受体胞内部分磷酸化的单克隆抗体。应用磷酸化蛋白的非特异性抗体,可以检测出EGFR蛋白的总量。由于EGFr在皮肤中高度表达并具有活性,皮肤非常适合用作该药的替代组织。在II期推荐的用药剂量下,用药期间获得的所有皮肤活检标本中,EGFR自身磷酸化都受到了抑制。在单药的II期试验中,少数患者中观察到客观缓解和疾病稳定期延长。由于没有检测肿瘤组织EGFR的表达或活性,肿瘤中的EGFR的活性是否被抑制还不清楚。这一点在整个EGFR靶向治疗类药物的研发中至关重要。如果能够通过EGFR酪氨酸激酶的残余活性明确区分无效者和有效者,就可以证明EGFR为治疗的靶点。优化作用于EGFr的药物可以克服无效者中所见的耐药问题。反之,如果肿瘤中的EGFR酪氨酸激酶的活性受到相同程度的抑制,那么这就不是有效的靶点。这种情况下,对于仅仅抑制EGFR酪氨酸激酶的药物,就无须进行进一步研究了。
3. 标准的II期临床试验设计
3.1. 单一疾病的单臂研究
传统的II期临床试验的研究策略是评价组织学类型明确且器官特异的肿瘤患者的客观缓解率。临床相关因素一般用于选择研究人群,针对某个肿瘤类型的依据一般是经验性,并以类似患者既往的标准治疗的缓解率作为参考。人为确定一个临床上与标准治疗有显著差异的目标缓解率,且错误拒绝一个有效治疗或接受一个无效治疗的概率是固定的。一般情况下,接受假阳性或假阴性结果的概率分别为5%和80-90%。这些参数决定了检出所期望的治疗作用所需要的样本量。假阳性结果可能导致对一个无效的治疗方案进行进一步临床试验,并被证实缺乏疗效。这样患者参加一个III期临床试验的时候,就把患者置于接受无效治疗而不是接受标准治疗的危险之中,同时还无谓地耗费了有限的资源。如果对一种药物在多个II期研究中针对不同的肿瘤类型进行评价,假阳性结果的累积概率就会增加。假阴性的II期试验则会导致放弃一种本来有可能成为有效治疗方案之一的药物。
为了尽量减少暴露于无效药物的患者的数量,目前,已经提出了达到总样本量之前,在一项研究中停止继续入选患者的统计学方案。Simon两阶段设计是此类规则中应用最广泛的。用标准治疗的缓解率作为下限,我们可以计算出最初的队列中检测到治疗效果所需要的病例数,并保证95%的把握排除劣效治疗的可能性。假如有充分的标准治疗的数据,则可以选择多种临床终点作为II期临床试验的主要终点。目前认为以无进展时间作为终点比采用客观缓解率更好,其优点是患者用一种新的治疗可能出现比较长的疾病稳定期,而按传统的缓解标准判断可能会忽视了这种受益。
4. 新的II期试验设计
4.1. 同情给药(Compassionate Use)或扩大用药(Expended Access)的II期试验
虽然很罕见,但有时也可能根据II期试验中令人信服的数据使药物获得批准。STI571治疗化疗失败的胃肠道间质瘤就是一个例子。目前肿瘤学方面药物审批体系的基础是III期试验中具有生存优势。快速完成上述研究存在实际操作上的限制,导致了很有前景的药物迟迟不能在难治性恶性肿瘤中应用。因此,FDA出台了一项条款,可以扩大仍处于III期试验当中的药物的临床应用。对于已经证明一线治疗可以延长生存期但后续治疗没有生存期受益的疾病,可以通过同情给药的II期试验来提供有前途的试验药物。采用这种方法前,必须有传统II期临床试验证明试验药物明显优于现有的治疗。如用多西他赛作为转移性乳腺癌的三线治疗(25)以及ZD1839对非小细胞肺癌的三线治疗都采用了这种研究方式。严密的毒性监测是进行此类研究的先决条件,但是,一般不强求进行严格的、相关的科学研究。在等待注册试验的关键性结果时,扩大的II期试验可以提供更多的毒副作用信息,可以对临床疗效进行更细致的估计。
4.2. 随机化II期试验
采用前面所述的药效学方法,可以确定靶向治疗的一个最小生物学有效剂量(minimum biologically effective dose,MBED)(图2)。这个剂量可能大大低于MTD。如果试验药物的作用机制是剂量依赖性的,而且没有进行生物学有效剂量评价,那么不同剂量研究药物的临床疗效可能不同。并且,独立的I期试验可能没有发现给药方案对药效学反应的影响。随机化的II期研究可以对上述假设进行检验。在研究设计中,2个或多个给药剂量或给药方案可以按照配对研究的方式进行比较。只要有足够的样本量,就可以检测出疗效的差异。
以贝伐单抗(bevacizumab,Roche)为例,在随机化的II期试验中,该药在MTD时的临床疗效优于MBED时的临床疗效。贝伐单抗是一种单克隆抗体,它与循环中的血管内皮生长因子-1(VEGF-1)结合,可导致目标蛋白的降解。单次用药的剂量递增试验显示,在用药剂量≥3mg/kg时,循环中检测不出VEGF。虽然没有按剂量依赖性的方式出现剂量限制性毒性,但用药量达到10mg/kg时即停止了剂量递增。作为I期研究中的药效学终点,即贝伐单抗用药后短期内血清VEGF浓度,不一定反映药物对VEGF受体家族信号传导的影响。由于上述原因,随机化的II期试验中,在110例晚期肾细胞癌患者中对3mg/kg、10mg/kg和安慰剂三组的疗效进行了比较。与3mg/kg剂量组和安慰剂组比,10mg/kg剂量组的无进展时间显著延长。这些结果提示,I期试验中所用的药效学终点不能预测临床受益情况。本例中,MBED的临床疗效与MTD相关的临床疗效有显著差异。
除了评价多种剂量以外,随机化的II期试验可以同时评价多个药物。这种情况下,所有治疗组的患者入选标准必须一致,但治疗可以不同。因为研究没有足够的把握度进行直接比较,所以II期研究的目的是探索疗效,而不是在试验药物和标准治疗之间做出决定性的比较。这些研究一般规模不够大,因此,不能根据生存期这样的终点来推断相对的优效性或劣效性。但是,这样的设计可为选择比较有前景的药物,以及优选进入III期试验的药物提供依据。一个II期临床试验可以有把握地对治疗方案的药效学终点的优效性做出推断,因此,II期临床试验可以包括一个标准治疗组,目的是检测出替代终点的差异。

图2. 随机化II期试验的设计
这种设计的主要局限性在于可检出有临床意义的疗效差异的统计学把握度。通常在III期临床试验开始前计算的样本量取决于单臂设计的II期临床试验中的患者的无进展时间和总生存期。II期试验中的事件发生率和所期望的疗效程度决定了有充分把握检测出治疗受益所需要的病例数。I期研究由于样本量小以及研究人群的不均一性,所以无法提供此类数据。因此,随机化II期试验的样本量估计主要取决于历史对照中的数据,这就会导致样本量估计的随意性,并且常常不够恰当。把握度不足的随机化II期试验可导致大量资源的损失和假阴性结果。这种设计的等效结果常常支持对最小剂量做进一步评估。如果对样本量估计过低导致对等效性的错误推断,那么随后的III期试验由于给药剂量不足而注定会失败。
4.3. 随机化停药设计
正在研发中的几个靶向治疗药物已经在临床前模型中表现出抑制细胞生长的作用,预计它们会通过延长疾病稳定期而不是肿瘤消退提供临床受益。在这类药物的II期临床评价中,无疾病进展时间可能是最有意义的临床终点。但是,不治疗的肿瘤也会有短时间的疾病稳定期。这就限制了将单组试验中的无疾病进展时间进行外推。随机化停药设计可以区别治疗相关的疾病稳定和自然病程所致的疾病稳定(图3)。研究初期所有患者都用试验药物治疗,在经过规定的疗程之后,评价疾病的客观缓解率。假定完全缓解和部分缓解的患者获得了治疗益处,则将继续用试验药物治疗,直到疾病进展为止。任何时间只要出现肿瘤进展就停止治疗。最初评价时疾病稳定的患者被随机分组,继续用试验药物或者停用试验药物。为了避免两组患者治疗方面的差异所产生的偏倚,采用双盲法给停药组的患者使用安慰剂。对两组病人进行随访,直到疾病进展为止。如果治疗组无疾病进展时间相对延长则表明疾病稳定是使用试验药物的结果。这些数据可以支持将抑制细胞的药物加入到III期临床试验中。
4.4. 药效学终点
按照客观缓解率设计的II期试验的把握度可能对试验药物的生物学活性估计过低。由于2.4.1.小节下所讨论的原因,许多靶向治疗可能会影响疾病进展而不诱导影像学可见的肿瘤消退。在这些情况下,通过相关性研究评价药效学作用就特别重要。为了能检测出临床缓解指标不明显,但其生物学活性显著的情况,必须以药效学终点设计II期临床试验。要实现这一目标有几个障碍,即检测方法在技术上必须对大多数患者是可行的;在连续多次进行肿瘤或替代组织活检的情况下,必须根据获得靶组织的难易程度来选择患者。外周血单核细胞和其他血液标志物具有最大优势,因为取得这些标本的可行性不是问题。还有,检测方法必须稳定。这就是说,个体自身和个体之间的变异不能太大。如果是将这种检测方法作为计算II期试验的样本量的依据,则必须在I期研究中实践这些检测方法。
检测肿瘤组织的主要局限性是要以安全的方式从有意义的病例数中获得标本。因此,相比而言,检测替代组织在可行性方面具有更大的优势。但是,不能假定药物在肿瘤的基因型和表型不同的组织中,其药效学的反应是相同的。分子影像学正在成为评价肿瘤微环境改变的一种方法。正电子发射断层照相术(PET)正在不断地改进用于这一目的。PET一般是以18F-氟-2-脱氧-D-葡萄糖(FDG)作为分子示踪剂,取患者的血标本,与FDG混合,然后重新注入患者体内。细胞摄取FDG和未标记葡萄糖的情况与代谢活性成比例。肿瘤细胞摄取的比例失调,导致FDG的浓度较高。随着放射性核素的衰变,发射出正电子,这些正电子可以用放置在患者周围的传感器检出。
FDG能够检测出治疗引起的代谢改变。如果一种治疗方法能够诱导细胞死亡,那么FDG-PET将可以检测出这种效应,且速度比传统的影像学方法(如计算机断层扫描)快得多。STI571治疗胃肠道间质瘤患者的II期研究已证明了这一点。这一发现的理论价值在于可以早期发现治疗无效的患者,使其换用其他治疗。FDG-PET不能提供有关治疗药物对分子靶点的影响的信息。为了评价其他化合物的摄取情况,正在研发其他的示踪剂。血流灌注和血管通透性对抑制血管生成的药物来说有重要意义,15O-水作为肿瘤灌注和血管通透性的标志物的研究正在进行中。
更加特异性的分子改变可以通过标记自身或药物配体来成像显示。最近发现的一种雄激素受体的自然配体5α-二氢睾酮可以用这种方法成像。已知前列腺癌细胞可以在体内外过度表达雄激素受体,非雄激素依赖性的前列腺癌细胞可以摄取16β-18氟-5α-二氢睾酮类,因此应用这种化合物对转移癌患者进行成像检查可以发现活动性病变。目前正在验证应用这种影像学方法评价可下调受体表达的治疗方法的疗效的可行性。类似策略可对任何导致配体受体下调的药物进行药效学评价。选择性雌激素受体修饰剂氟维司群(fulvestrant),也是用于进一步研究的理想的候选药物。虽然氟维司群的直接作用机制是下调雌激素受体,但其下游的许多化合物可导致细胞表面、细胞浆和核受体的表达降低。这类药物非常适合进行分子影像学研究。
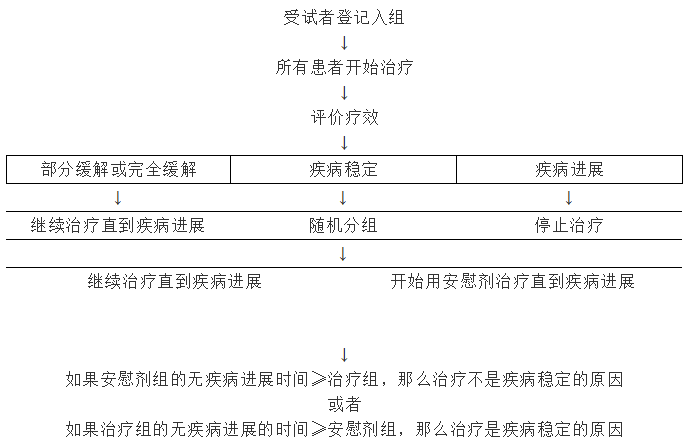
图3. 随机化停药设计
5. II期试验中的个体化治疗
开发稳定的药效学检测方法为在II期试验中针对个体患者调整治疗提供了基础。人群中药代动力学的变异会造成某些患者的给药方案不当。在药物清除速度快的患者中,所用剂量可能低于治疗剂量,这样的给药方案可能导致药物暴露量不足。肿瘤方面的因素也可以导致具有相同组织学亚型和分子谱型的肿瘤患者中药效学反应的差异。这些可能反映在替代组织中相同分子靶点的药效学作用上,但也可能反映不出来。在这层意义上,替代组织最多是效应的标志物,而不是预测指标。
对于分子靶向治疗来说,生物学有效剂量的范围可能很宽。贝伐单抗的MBED和MTD之间的差异至少有3倍。在I期试验中,剂量没有递增到MTD,所以两者间的差异可能更大。停止剂量递增的主要原因是剂量≥3mg/kg时已经完全清除了循环中的VEGF。血清VEGF水平的恢复与剂量有关,剂量越大,VEGF水平的恢复时间越长。剂量为3mg/kg时开始检测不到VEGF,因此被认为是MBED。这个剂量和I期试验中评价的最高剂量10mg/kg都被用到了II期试验中。在肾细胞癌患者的随机化II期试验中,对主要终点无疾病进展时间的评估中10mg/kg剂量组优于3mg/kg剂量组。这个结果与生物学有效剂量的定义产生了矛盾,提示检测不出VEGF水平不是代表药物作用机制的终点,而VEGF被耗尽所用的时间,可能是预测指标。这个假设可以在II期试验中通过剂量调整进行检验。
所涉及的剂量调整方法要对药效学进行实时分析。贝伐单抗的例子很有启发意义,因为这种检测方法可以在所有患者中进行,而且比较容易实现。正在进行中的III期试验中贝伐单抗每2周用药1次。可以设计一个随机化的II期试验,将患者随机到MBED、MTD或者通过药效学确定的调整剂量组中的任一组。在第3组中,患者开始按MBED治疗,因为如果毒性是剂量依赖性的但活性不是剂量依赖性的,则最好用最低剂量。在第1个疗程中,按一定的间隔时间评价药效学终点。达到目标药效学作用的患者继续治疗,不调整剂量。没有达到目标效应的患者,下个疗程增加剂量。这类研究能够识别易于达到药效学目标的治疗策略。如果MTD组和其他两组之间药效学一致,则提示在III期试验中应当采用更小的剂量,因为这样可以尽量减少剂量相关的副作用。
一项有3个对照组、随机化II期试验所要求的样本量比只进行客观缓解率比较的单组II期试验所需要的样本量大。但是,对II期试验的投入将优化III期试验中治疗方案的选择。对于临床疗效与标准治疗疗效无显著性差异时,只有那些具备明确药效学作用的药物才会进入下一步研发。如果在II期试验中没有对剂量、给药方案和药效学终点进行充分研究,进入III期试验的方案可能不是最佳的给药方案,则III期试验失败的风险就比较大。在失败的III期试验完成以后,很难对这些参数做出改变。
由单一致病因素导致疾病发生的人类肿瘤很少见。仅仅在这种情况下,单一靶向治疗能够大大改变肿瘤的自然病程,因此,慢性髓性白血病和胃肠道间质瘤对STI571的敏感性不能推广到其他多数肿瘤的治疗中。越来越多的证据表明,靶向治疗药物的联合应用可以产生更深远的抗癌作用。如果为了在标准治疗的基础上增强疗效,需要同时针对多个致病因素,因此可以用靶向药物联合治疗,尽管这些靶向药物的单药疗效很低,但对于联合治疗是必不可少的。然而, II期试验中继续评价药物对其靶点的影响至关重要。如果药物未能显示出充足的单药活性以支持在III期试验中与标准治疗进行直接比较,就应当依据研究中互补作用机制的临床前证据,继续与其他药物按联合治疗进行评价。
参考文献:
1.CalvertAH,Newell DR,Gumbrell LA,et al.Carboplatin dosage:prospective evaluation of asimple formula based on renal function.JClin Oncol 1989;7:1748-1756.
2.EttingerDS.Evaluation of new drugs in untreated patients with small cell lungcancer:Its time has come.J Clin Oncol 1990;8:374-377.
3.LandysK.Mitoxantrone as a first-line treatment of advanced breast cancer.Invest New Drugs 1985;3:133-137.
4.HendersonIC,Allegra JC,Woodcock T,et al.Randomized clinical trial comparing mitoxantronewith doxorubicin in previously treated patients with metastatic breast cancer.J Clin Oncol 1989;7:560-571.
5.MoertelCG,Schmitt AJ,Hahn RG,et al.Effects of patients selection on results of phaseII chemotherapy trials in gastrointestinal cancer.Cancer Chemother Rep1974;58:257-260.
6.KarnofskyDA,Burchenal JH.The clinical evaluation of chemotherapeutic agents incancer.In:Macleod CM,ed.Evaluation ofChemotherapeutic Agents.New York:Columbia University Press,1949.
7.AlbaE,Asstus R,de Andres L,et al.Anticipatory nausea and vomiting prevalence andpredictors in chemotherapy patients.Oncology1989;46:26-30.
8.RobertJ,Hoerni B.Age dependence of the early-phase pharmacokinetics of doxorubicin.Cancer Res 1983;43:4467-4469.
9.GravesT,Hooks MA.Drug-induce toxicities associated with high-dose cytarabinearabinoside infusions.Pharmacotherapy1989;9:23-28.
10.MooreES,Faix RG,Banagale RC,Grasela Th.The population pharmacokinetics oftheophylline in neonates and young infants.JPharmacokinetics Biopharm 1989;17:47-66.
11.GraselaTH Jr,Donn SM.Neonatal population pharmacokinetics of Phenobarbital derivedfrom routine clinical data.Dev PharmacolTher 1985;8:374-383
12.HuntCM,Westerkam WE,Stave GM.Effect of age and gender on the activity of humanhepatic CYP3A.Biochem Pharmacol1992;44:275-283.
13.KalowW.Ethnic differences in drug metabolism.ClinPharmacokinet 1982;7:373-400.
14.LiuHJ,Han CY,Liu BK,et al.Ethinic distribution of slow acety lator mutations inthe polymorhphic N-acetyltransferase(NAT2)gene.Pharmacogenetics 1994;4:125-134.
15.MarsoniS,Hoth D,Simon R,et al.Clinical drug development:an analysis of phase IItrials,1970-85.Cancer Treatment Rep1987;71:71-80.
16.StaquetMJ,Byar DP,Green SB,et al.Clinical predictivity of transplantable tumor systemsin the selection of new drugs for solid tumors:rationale for a three-stagestrategy.Cancer Treatment Rep1983;67:753-756.
17.ShoemakerRH,Wolpert-Defilippes,Kern DH,et al.Application of a human tumor colony-formingassay to new drugs screening.Cancer Res 1985;45:2145-2153.
18.ShoemakerRH,Monks A,Alley MC,et al.Development of human tumor cell line panels for usein disease-oriented drug screening. ProgClin Biol Res 1988;48:265-286.
19.AlleyMC,Soudiero DA,Monks et al.Feasibility of drug screening with panels of humantumors cell lines using a microculture tetrazoium assay.Cancer Res 1988;48:589-601.
20.PeckCC,Barr WH,Benet LZ,et al.Opportunities for integration ofpharmacokinetics,pharmacody-namics,and toxicokinertics in rational drugdevelopment.Pharm Res 1992;9:826-833.
21.WorkmanP,Graham MA.Cancer Surveys 18(Pharmacokinetics and Cancer Chemotherapy),1993.
22.SheinerLB,Ludden TM.Population pharmacokinetics/dynamics.Annu Rev Pharmacol Toxicol 1992;32:185-209.
23.WhitingB,Kelman AW,Grevel J.Population pharmacokinetics:theory and clinical practice.Clin Pharmacokinetics 1986;11:387-401.
24.SheinerLB,Beal SL.Bayesian individualization of pharmacokinetics:simple implementationand comparison with non-Bayesian methods.JPharm Sci 1982;71:1344-1348.
25.KruijtzerCMF,Verweij J,Schellens JH,et al.Docetaxel in 253 previously treated patientswith progressive locally advanced or metastatic breast cancer:results of acompassionate use program in The Netherlands.Anticancer Drugs 2000;11:249-55.
