发稿时间:2019-03-23 来源:中国临床药理学杂志
摘要:目的 对高变异药物,在进行参比制剂标定的生物等效性试验(RSABE)设计时,就例数的选择提供一种估算方法。方法 建立了数据模拟的方法,按照参比制剂标定的计算方法,估算在不同几何均值比值(GMR)、个体内变异(CVw)的情况下,达到一定的把握度时所需要的样本量。结果 通过模拟计算,获得了在几何均值比值0.85~1.20,个体内变异30%~80%,在80%和90%把握度下所需要的例数。结论 所建立的数据模拟方法求算的例数结果可用于开展RSABE的例数设计。针对高变异药物,使用参比制剂标定的三交叉试验可显著提高研究的效率。
关键词:高变异药物;参比制剂标定的平均生物等效性;样本量;把握度
在生物等效性研究中,高变异药物的生物等效性研究可占总数量的10%左右。对这些药物的研究设计,可以采用目前常规的2周期2序列、4周期2序列交叉的平均生物等效性(ABE)研究方法,也可采用参比制剂标定的生物等效性研究(RSABE),其研究设计包括参比制剂重复的三交叉试验(又称部分重复试验)和四交叉试验(又称完全重复试验)。常规的生物等效性研究方法是评价使用两种制剂后,其所评价的药代动力学评价参数的几何均值比值(对数转换后的差值)是否在允许的范围内。目前,在国际上,这个允许的判断区间(界值)大多是80%~125%。当个体内变异很高时,如达到30%以上(高变异药物的定义范围)时,要满足以上的条件来获得等效的结果,需要很大的研究例数才可以完成,这会带来很大的资源消耗和受试者不必要的药物暴露。为此,在评价高变异药物时,欧洲药品管理局(EMA)和美国食品药品监督管理局(FDA)相继提出了参比制剂标定的生物等效性研究方法。在这些方法中,等效性界值以参比制剂个体内变异的而定。当然,EMA和FDA的方法之间还存在一定的差异。EMA方法所针对的界值调整主要针对达峰浓度(Cmax),且放宽到一定范围时就不再放宽;而FDA的方法是对Cmax和血药浓度-时间曲线下面积(AUC)均进行放宽,且没有限度要求。在RSA-BE的试验设计中,因不考虑受试制剂的个体内变异,参比制剂重复的三交叉试验是进行高变异研究时研究效率最高的试验设计。对于可能是高变异的药物,为开展三交叉、四交叉RSABE的试验,需要首先估算其样本量的大小和把握度。对于二交叉ABE的试验设计,有多篇文献和教科书报道了样本量和把握度的估算方法。但对三交叉和四交叉的RSABE试验设计,关于样本量的大小估算,文献报道很少。为了了解三交叉、四交叉研究设计的数据特点,以FDA规定的RSABE方法为依据,用数据模拟的方法进行三交叉和四交叉RSA-BE试验的例数和把握度估算,为开展此类研究提供帮助。
1 参比制剂标定的生物等效性研究原理
在RSABE研究中,同样也是根据评价的药代动力学参数(如Cmax,AUC)在对数转换后的差值(μT-μR)的大小做出是否等效的结果判断,但其置信区间的大小考虑了参比制剂的个体内变异。可以通过以下公式进行计算。
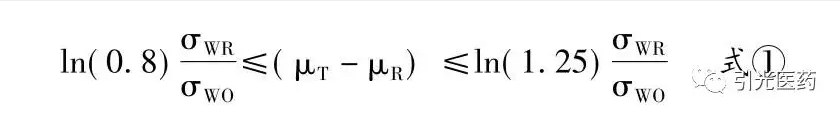
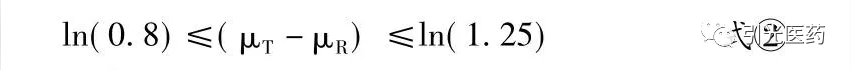
式②即为常规的ABE计算公式。
对于σWO的取值,FDA规定为0.25,但FDA同时也规定,RSABE方法只能在参比制剂个体内变异(σWR)>0.294时方可以使用,低于此值时,仍然需要使用常规的ABE方法。根据换算公式,对数转换后的σWR>0.294对应于对数转换前的CVW=0.30。
2 RSABE试验样本量的计算方法
2.1 模拟数据集的产生
在三交叉和四交叉试验中,对于受试制剂,用于等效性判断的某一参数在对数转换后可以由群体均值(σT)、个体内变异(σW)、个体间变异(σB)、周期间变异(π)和序列变异(γ)组成。其中个体间变异为随机因素,符合均值为0、方差(σB2)的正态分布;个体内也为随机因素,符合均值为0、方差σW2的正态分布;周期间变异和序列变异一般用固定因素来描述。本文中,为了简化起见,周期间变异和序列变异暂不加入。对同一个体,在不同周期中,其个体间变异保持不变,但其个体内变异,在不同周期间是不同的。对于参比制剂,其均值是μR,可根据μT-μR=ln(θ),求得的值μT(为受试制剂和参比制剂几何均值的比值)。另外假设参比和受试制剂的个体内方差σWR2=σWT2。依据上述原理,通过计算软件,可以模拟产生所需要的三交叉、四交叉试验数据,依此数据进行三交叉、四交叉的RSABE计算。每种条件下,每种序列、每个周期产生10万个数据,3种序列、3个周期产生90万个数据用于模拟数据的计算;2种序列、4个周期产生80万个数据用于模拟数据的计算。
2.2 模拟数据集的抽样
在实际试验时,常常按照均衡设计的方式进行试验,因此,模拟数据抽样时,从每个序列中抽取相同数量的受试者,组成抽样数据。对于三交叉试验,因存在3种序列,组成的受试者例数为3的倍数;对于四交叉试验,只有2种序列,组成的受试者例数为2的倍数。考虑到产生的数据量足够的多,在实施每次抽样时,受试者不进行替换。
2.3 模拟数据集的计算
根据FDA提供的计算公式,计算一定例数情况下,每次抽样后获得的结果,每种条件重复2000次,汇总成功率。当抽样数据计算所获得的σWR<0.294时,仍然采用ABE的方法进行计算和判断。
当重复的次数足够多时,其汇总的抽样成功率即为把握度,计算把握度为80%和90%时所需要的例数。
3 结果
通过反复抽样,计算所获得的参比制剂个体内变异和设计时的变异大小是一致的,提示计算过程无误。
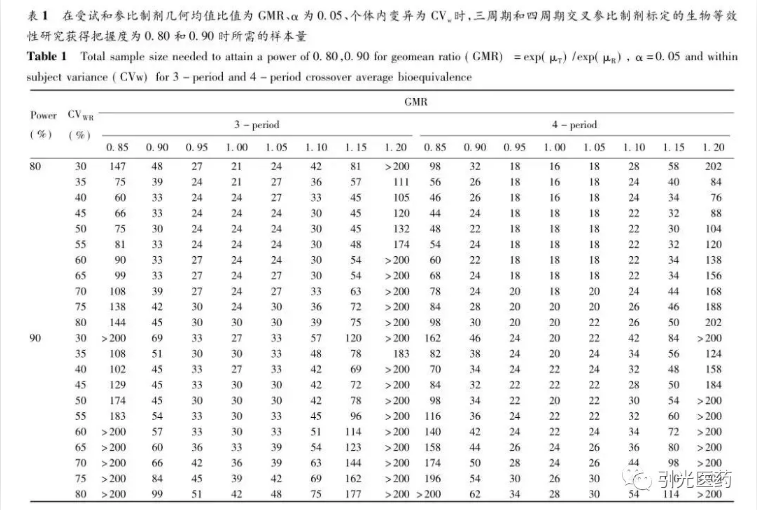
表1汇总了不同把握度、CVWR和θ值时的三交叉和四交叉受试者例数。由于篇幅的限制,没有提供更细致的数据结果。
4 讨论
本文通过数据计算,获得了在不同条件下所需要的研究例数,所获得的例数和把握度数据可以用于三交叉和四交叉的RSABE设计。从所提供的结果看,RSABE方法可以大大减少研究的例数,体现了这种设计的优势。
在通过数据模拟的方法获得了研究结果后,作者也查阅到参考文献报道的结果。在所提供的文献中,受试者的例数并不是3和2的倍数。通过对比,本文所获得的结果和文献结果,在数据重叠的部分中,两者是一致的,说明本文所得到的结果是可靠的。
在本文的例数估算中,本课题组假设参比制剂和受试制剂具有相同的变异,但实际上,会存在不一致的情况。参比制剂的个体内变异对对数转换后的差值(μT-μR)有影响,模拟数据结果显示:当受试制剂的个体内变异大于参比制剂时,本试验中确定的例数需要考虑上调,才能获得等效的结果。出现这种情况可能是由两种制剂的释放机制不一致所致,也可能是其他原因。相反,当受试制剂的个体内变异小于参比制时,失败率略有下降,如保持试验例数不变,把握度会提高。模拟结果显示,个体间的变异大小对失败率没有影响。
RSABE设计因其设计原理不同,不能简单套用ABE例数的估算方法。从本文的计算结果中可见,随着受试者个体内的变异加大,入选受试者的例数并不加大太多,这主要是参比制剂校正的试验特点,放宽了接受的限度。
另外,FDA法规还规定对于使用RSABE的研究设计,需要设定最低例数不能低于24例,这样表1中的最低例数不能低于24例,以避免RSABE的研究设计被不当使用。
