发稿时间:2019-04-09 来源:南京医科大学学报
作者:姜慧勇1,2,娄冬华1*(1南京医科大学公共卫生学院,江苏南京211166;2科文斯医药研发(北京)有限公司,北京100015)
来源:南京医科大学学报(自然科学版) 第38卷第10期 2018年10月
关键词:CDISC;既往病史;不良事件;合并用药;逻辑核查;数据清理
临床试验数据管理中,病例报告表(case report form,CRF)中的既往病史(medical history,MH)、不良事件(adverse event,AE)和合并用药(concomitant medication,CM)的数据没有普遍规律可循,大量文本型数据需要研究者填写,数据量较大,一般需要有相关医学背景知识人员才能完全清理等特点,是数据清理的难点和重点。这部分临床试验数据的清理是衡量一个临床试验项目数据质量的关键点,也是监管机构评价临床试验真实、可靠、完整的切入点。基于当前业内临床试验数据管理趋势,本文欲以临床数据交换标准协会(clinical data interchange standards consortium,CDISC)⁃临床数据采集整合标准(clinical data acquisition standards harmonization,CDASH)为基础,探索目前临床试验MH、AE和CM的CRF表设计的成熟性和规范化程度。建立一套通用、广覆盖的MH、AE和CM数据逻辑核查方案(EDC自动核查、SAS程序核查),用以清理MH、AE和CM三者普通逻辑错误和数据彼此矛盾的情况。从而标准化、模块化临床试验前期数据管理的准备工作,降低试验成本,提高效率及数据质量。
1 基于CDISC⁃CDASH标准的MH、AE和CM的CRF表设计
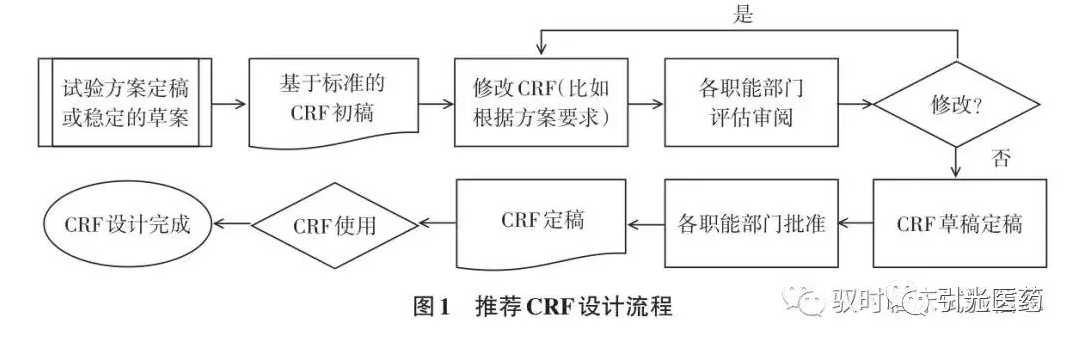
1.1 CRF设计流程
CDISC建立了涵盖研究方案设计、数据采集、分析、交换、递交等环节的一系列标准。其中CDASH描述了基础数据采集域和CRF标准问题文字描述的变量、实施指南和最佳操作方案的融汇。尽管CDASH涵盖了MH、AE和CM 3个表格最通用的数据采集要求,可以应用于绝大多数临床试验,但是由于每个临床项目方案不同,所有CRF表格还是需要进行独立的设计评估流程(图1)。
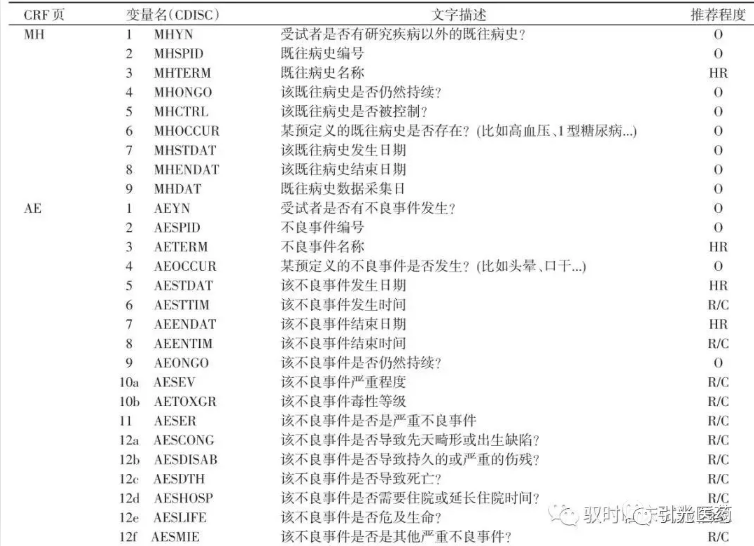
1.2 MH、AE和CM的CRF表
CDASH将所有需要采集的变量分为3类(表1),强烈推荐:一般情况下CRF都要包含这些变量;推荐:特定条件下需要包含这些变量;可选:根据方案或申办者实际需求选择性包含这些变量。除此之外,也可根据方案的实际情况,添加其他变量。

1.3 CRF表设计的注意点
除了事件名称外,尽量避免开放性的文本框。目前绝大多数临床研究都会使用MedDRA和WHO Drug分别对AE、MH和CM的名称进行编码,从而使后期数据统计分析变得更加便捷。减少开放性文本框,意味着研究者填写更加便利清晰,数据质疑的数量将会大大减少。
使用通用术语来定义CRF采集变量的回答选项。不同研究的数据采用相同变量名、回答选项术语代码、结构或格式,有助于数据分析和交流,也方便了向药政管理部门提交。
具体化想要得到的答案,充分考虑可能存在的回答。比如合并用药开始日期可能存在不完全的情况,在设计阶段中,“日”需要增加“UN”选项,“月”需要增加“UNK”选项,整体日期需要增加“Unknown”选项。
2 逻辑核查
逻辑核查是指通过计算机编程,利用计算机软件的功能,对已录入CRF的数据进行验证,以发现存在问题的数据。CDISC中对逻辑核查的定义:对预期的内容逻辑、数据字段格式、范围或其他属性进行一个可追溯的、自动化的评估过程。逻辑核查的优点:可以对录入CRF的数据进行实时反馈;及早发现前后数据矛盾点;提高数据质量,使监查员和数据管理员有更多精力放在更为复杂的数据清理上。
2.1 逻辑核查的分类
根据目前普遍的数据管理流程,逻辑核查可分为系统实时逻辑核查和SAS程序逻辑核查两类。同时按照核查数据点的不同,又可分为单一变量逻辑核查和多变量交互逻辑核查。
2.2 逻辑核查的流程
按照计算机程序的开发过程,逻辑核查的步骤包括:逻辑核查需求定义(edit check specification,ECS)、开发、测试、应用及变更控制。其中尤以ESC的设定最为重要,决定了数据清理的质量和效率。
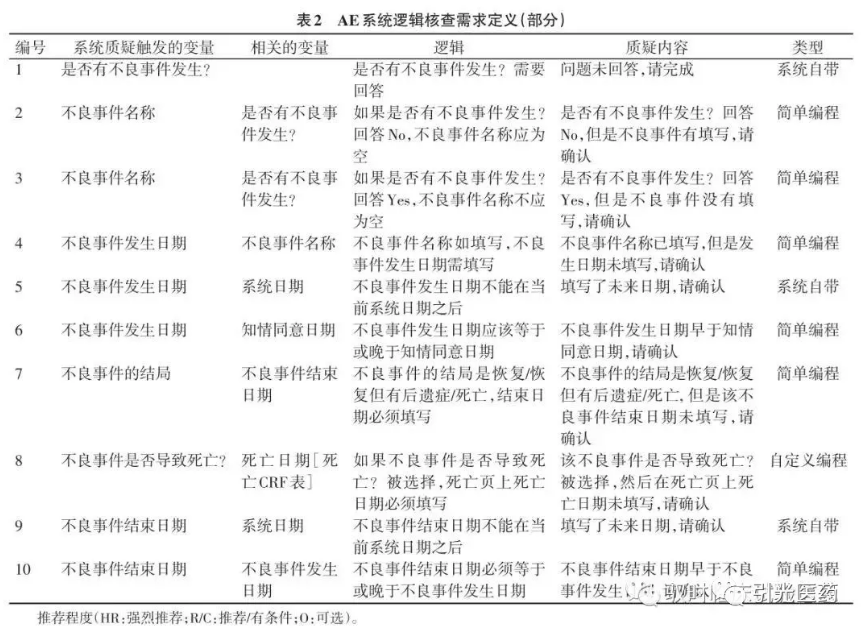
2.3 MH、AE和CM的CRF表的系统实时逻辑核查
在设计ESC时需要包含以下内容(以AE表举例):逻辑核查的访视名、系统质疑触发的变量、相关的变量、逻辑、质疑内容和逻辑核查类型(表2)。
2.4 MH、AE和CM的CRF表的SAS逻辑核查
通常来说SAS逻辑核查相对系统实时逻辑核查要复杂许多,往往是几个表中的多个变量交互核查,有的甚至需要借助其他相关资料。比如表3是一条核查抗糖尿病用药与MH和AE逻辑关系的ECS,该条逻辑核查借助了糖尿病相关药品的whoDD编码列表(526条,由医学专家和编码人员共同提供)和糖尿病相关事件MedDRA编码列表(80条,由医学专家和编码人员共同提供)。SAS程序员在获取MH、AE和CM数据库后,结合上述编码列表,通过SAS编程将不符合逻辑的合并用药全部列出,由数据管理员进行核查并发出质疑。

3 结束语
尽管我国临床试验数据管理起步相较于欧美等发达国家较晚,但随着我国国家食品药品监督管理总局对数据质量的日益重视,数据标准化是大势所趋。MH、AE和CM的数据清理只是临床试验数据管理的一部分,更多其他类型的病理报告表有待统一完善,从而帮助我们更高效、高质地开展试验。
