发稿时间:2020-05-08 来源:西格玛医学
前言:2017年、2018年我们发布了医疗器械人类遗传审批流程方法及解读,伴随着2019年7月1日《中华人民共和国人类遗传资源管理条例》(国务院令第717号)实施,这些老的帖子都已经不适用。为此我们写了一个医疗器械和IVD遗传办申报的流程【2020年版】 。
主要分为三个部分:
1、遗传办发展历史
2、遗传办申报分类
3、申报适用情况探讨
第一部分:遗传办发展历史
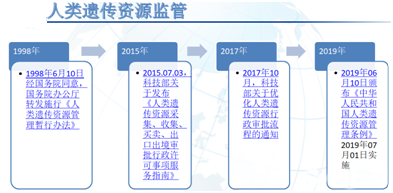
1998年,国务院办公厅转发了科技部、卫生部制定的《人类遗传资源管理暂行办法》(国办发[1998]36号)(以下简称《暂行办法》)。2015年3月,根据工作发展的需要,中央编办将依据《暂行办法》设定的原“涉及人类遗传资源的国际合作项目审批”行政许可名称和审批任务变更为“人类遗传资源采集、收集、买卖、出口、出境审批”。开始的时候,主要涉及科研领域,随后拓展到临床试验领域,随着科技部,科技厅的年次批次检查,企业、CRO、药物临床试验机构也俞发地重视。最初采用纸质递交和审批,2016年10月14日,科技部开通了网上申报系统。2017年10月26日,科技部发布了优化人类遗传资源行政审批流程,于2017年12月1日进行正式实施。2019年《中华人民共和国人类遗传资源管理条例》(国务院令第717号)实施,目前我们进行申报,全部按照此流程。这次又进行了更新,对采集、保藏、科学研究审批、材料出境、临床试验备案、信息对外提供,重要遗传信息申报和特定地区登记都进行了细化:(1)目前国际合作的上市前药物及上市后药物临床研究、科研类研究,医疗器械和IVD的临床研究都需要进行申报。(2)非国际合作,但例数大于500例的上市前药物及上市后药物临床研究、科研类研究,医疗器械和IVD的临床研究都需要进行申报。
申报指南种类如下图:

各种申报指南下载:
https://fuwu.most.gov.cn/html/jcxtml/20181218/2837.html?tab=fwzn
第二部分:遗传办申报分类
2.1 遗传资源如何分类?
人类遗传资源包括人类遗传资源材料和人类遗传资源信息。人类遗传资源材料是指含有人体基因组、基因等遗传物质的器官、组织、细胞等遗传材料。人类遗传资源信息是指利用人类遗传资源材料产生的数据等信息资料。任何国际合作试验都需要收集信息,导致所有临床试验和研究都需要申报,譬如非干预研究等,只要涉及收集信息。
2.2 申报流程如何进行分类?
从研究阶段、出料出境、临床试验例数、中心实验室采用等角度分析来看。医疗器械和IVD临床研究按照研究阶段或者目的,可以分为上市前临床研究和上市后临床研究,目前大部分医疗器械研究是上市前临床研究。在材料出境方面,以不出境为主;在例数方面,大于500例需要申报采集审批,医疗器械临床试验例数普遍小于500例,但一些上市后研究,或者冠脉支架及创新介入等产品,IVD三类上市前试验等通常会有超过500例的情况,需要在原有申报类型上,同时申报采集审批;药物临床试验采用中心实验室(central lab)较多,目前以国际合作科学研究审批为主,医疗器械不像药物,以院内采集和检测为主,基本可以走国际合作临床试验备案程序。对于一些骨科,介入类产品或者特殊检测血清银离子等才需要第三方阅片中心或检测所。经总结如下,仅供参考:
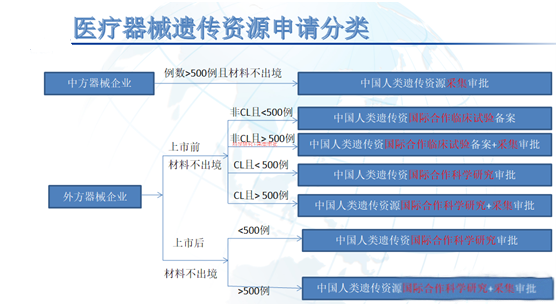
同时结合从医疗器械采用EDC系统,申报主体等进行分析。目前遗传办的遗传资源信息对外提供或开放使用备案,进行备案或审批后,再获取信息对外提供备案号,进行关联备案。申报主体方面,目前采集审批要求是必须研究中心(研究中心必须是公立医院,如是民营医院,不能有外资成分)申报,以组长单位申报为主流。如同时涉及国际合作科学研究审批,结合采集审批统一由研究中心申报,流程上操作应该更顺利。
第三部分:申报适用情况探讨
3.1 中国人类遗传资源采集审批
在老申报流程下,临床试验部分对采集没有明确要求。2019年7月后,对采集审批进行了细化,采集申报的范围为罕见病、具有显著性差异的特殊体质或生理特征的人群;规定筛选数量累积500人以上。既往临床研究没有500例的要求,几乎不涉及采集审批,但新条例通过500例数量将大部分临床试验扩入采集申报范畴,增加了临床试验遗传办申报的周期和费用。申报主体为中方单位,也就是医院。譬如国内IVD企业的三类产品基本都超过1000例,目前都必须申报采集审批,不管样本或者受试者是免知情还是签署知情。
3.2 国际合作临床试验备案
适用于为获得相关药品和医疗器械在我国上市许可,在临床机构利用我国人类遗传资源开展国际合作临床试验、不涉及人类遗传资源材料出境的情况。“在临床机构”包括:(一)所涉及的人类遗传资源仅在临床机构内采集、检测、分析和剩余样本处理等;(二)所涉及的人类遗传资源在临床机构内采集,由临床机构委托的单位进行检测、分析和剩余样本处理等。临床机构应与其委托的单位签署正式协议,明确委托检测和分析的人类遗传资源材料的种类、数量、检测内容、转运方式、剩余样本和数据信息处理方式等,并对其委托的活动负责。譬如何以体现是临床机构委托的中心实验室?是由临床试验机构与中心实验室签署合同,但目前启用的中心实验室,均为申办方与中心实验室签署合同,则不符合备案流程。西格玛医学认为,后续可以与研究中心探讨,将研究经费打入研究中心,由研究中心委托中心实验室,是否可以作为一种操作途径,作为审批转备案合规化操作的一部分,当然这也需要考虑申办方合规部门审查通过。
3.3 国际合作科学研究审批
国际合作科学研究审批是针对国际合作中临床试验备案不适用情况外的其他情况。只要开展国际合作研究,不备案,即需要审批。
3.4 人类遗传资源材料出境审批
对利用中国人类遗传资源开展国际合作科学研究,或者因其他特殊情况确需将中国人类遗传资源材料运送、邮寄、携带出境的情况,但必须是具有法人资格的中方单位申报,很少有临床试验需要材料出境。
3.5 信息对外提供或开放使用备案
将人类遗传资源信息向外国组织、个人及其设立或者实际控制的机构提供或开放使用。申请单位应为中方单位。申请人登录网上平台提交信息备份,并确定备份成功,获得信息备份号。信息备份成功后,申请人可登录网上平台在线提交备案材料,获得备案号。申请人获得备案号,即可将人类遗传资源信息向外国组织、个人及其设立或者实际控制的机构提供或开放使用。申办方在临床试验之前,需要考核EDC系统及数据管理的提供方及是否有外资成分。
综上所述,在临床研究中,需要考虑以上几种情况单独操作,或者综合操作。
文章来源于西格玛医学 ,作者西格玛医学
